![]()
![]()
Biochemical and biological functions of proteins are the product of both the overall fold of the polypeptide chain, and, typically, structural motifs made up of smaller numbers of amino acids constituting a catalytic center or a binding site that may be remote from one another in amino acid sequence. Detection of such structural motifs can provide valuable insights into the function(s) of previously uncharacterized proteins.
Technically, this remains an extremely challenging problem because of the size of the Protein Data Bank (PDB) archive. We have developed a new approach that uses an inverted index strategy capable of analyzing >200,000 PDB structures with unmatched speed. The efficiency of our inverted index method depends critically on identifying the small number of structures containing the query motif and ignoring most of the structures that are irrelevant. Our approach enables real-time retrieval and superposition of structural motifs, either extracted from a reference structure or uploaded by the user.
Structural motif searching is available as part of the RCSB Advanced Search and RCSB Mol* plugin. Help documentation is available.
Current benchmark times to search in 208,702 PDB structures and 1,068,577 AlphaFold/RoseTTAFold predictions as of 8/16/23, obtained on an instance with 6 cores and 64 GB memory. All structure data is held in memory, inverted index data is read from an SSD.
| Motif | Definition | Found Assemblies | 'Paths' Time [ms] | 'Score' Time [ms] | Total Time [ms] |
|---|---|---|---|---|---|
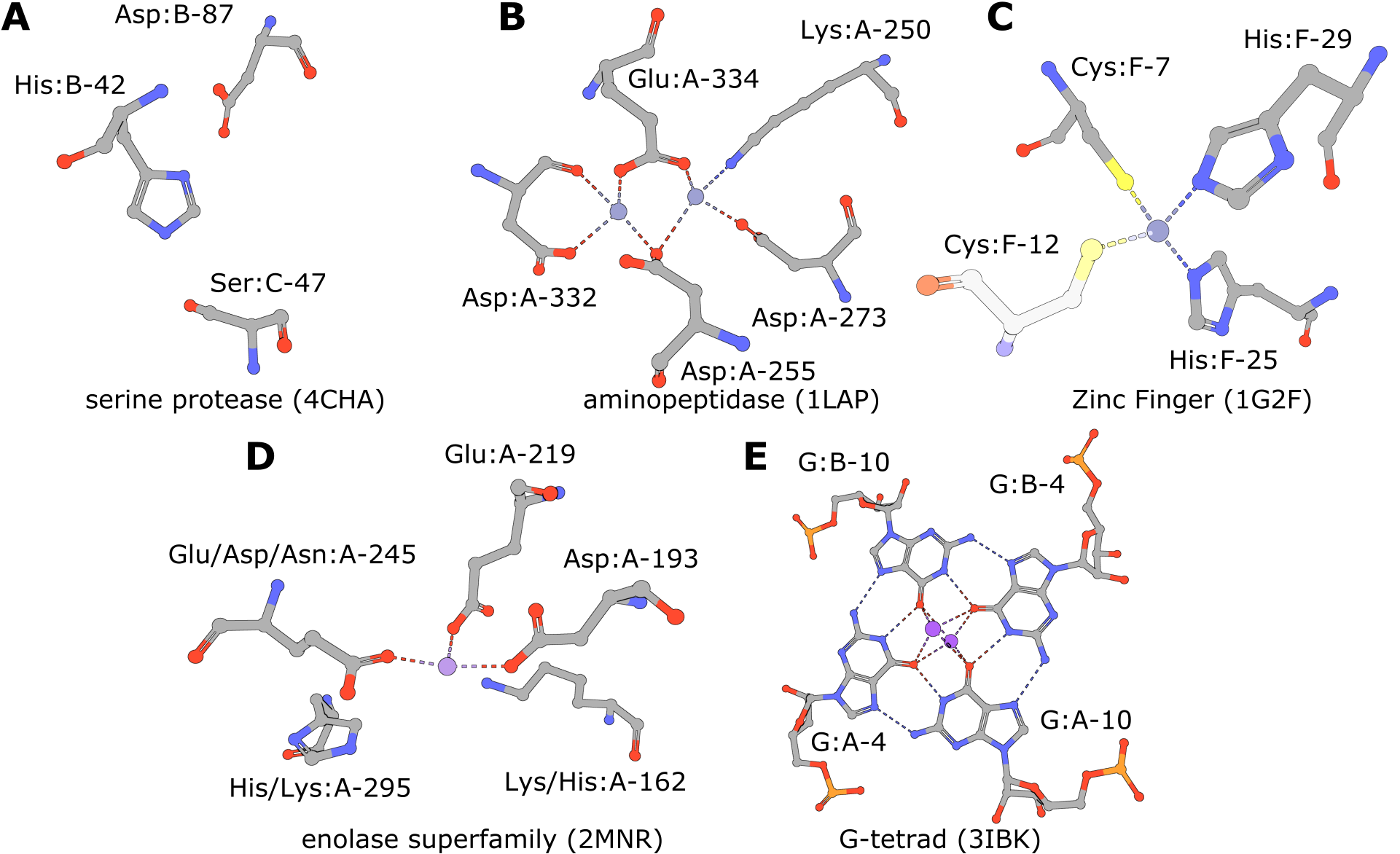
| Serine Protease | 4cha - His:B-42, Asp:B-87, Ser:C-47 | 5,309 | 618 | 22 | 673 |
| Aminopeptidase | 1lap - Lys:A-250, Asp:A-255, Asp:A-273, Asp:A-332, Glu:A-334 | 91 | 158 | 1 | 181 |
| Zinc Fingers | 1g2f - Cys:F-7 His:F-25 His:F-29 | 739 | 135 | 3 | 160 |
| Enolase Superfamily | 2mnr - Lys:A-162, Asp:A-193, Glu:A-219, Glu:A-245, His:A-295 | 192 | 253 | 2 | 275 |
| Enolase Superfamily (exchanges) | 2mnr - Lys/His:A-162, Asp:A-193, Glu:A-219, Glu/Asp/Asn:A-245, His/Lys:A-295 | 210 | 2,996 | 14 | 3,032 |
| RNA G-Quadruplex | 3ibk - G:A-4, G:A-10, G:B-4, G:B-10 | 85 | 2,364 | 236 | 2,622 |
| Motif | Definition | Found Assemblies | 'Paths' Time [ms] | 'Score' Time [ms] | Total Time [ms] |
|---|---|---|---|---|---|
| Serine Protease | 4cha - His:B-42, Asp:B-87, Ser:C-47 | 10,254 | 1,710 | 125 | 1,988 |
| Aminopeptidase | 1lap - Lys:A-250, Asp:A-255, Asp:A-273, Asp:A-332, Glu:A-334 | 647 | 352 | 7 | 389 |
| Zinc Fingers | 1g2f - Cys:F-7 His:F-25 His:F-29 | 9,442 | 492 | 92 | 686 |
| Enolase Superfamily | 2mnr - Lys:A-162, Asp:A-193, Glu:A-219, Glu:A-245, His:A-295 | 328 | 659 | 5 | 689 |
| Enolase Superfamily (exchanges) | 2mnr - Lys/His:A-162, Asp:A-193, Glu:A-219, Glu/Asp/Asn:A-245, His/Lys:A-295 | 350 | 5,246 | 25 | 5,296 |
| RNA G-Quadruplex | 3ibk - G:A-4, G:A-10, G:B-4, G:B-10 | 85 | 2,453 | 253 | 2,742 |
Search for all assemblies that contain hits with an RMSD <2 Å. 'Paths' refers to the time spent on inverted index operations, which identify all candidate structures that contain the motif. 'Score' refers to the time spent on aligning candidate structures to the query and computing RMSD values.
Computed structure models ignore unreliable regions with pLDDT <70.
- nucleotide support
- inter-chain & assembly support
- position-specific exchanges
- modified residues
- support for computed structure models, like from AlphaFold
- detect motifs in a structure of interest
strucmotif-search is distributed by maven and supports Java 11+. To get started, append your pom.xml by:
<dependency> <groupId>org.rcsb</groupId> <artifactId>strucmotif-search</artifactId> <version>0.22.0</version> </dependency>An alternative way to use the library is cloning this repository and building the corresponding Maven modules.
The Strucmotif class provides a fluent API to process structural motif queries.
Strucmotif.searchForStructures() // several ways can be used to define the query motif - e.g., specify a PDB entry id .defineByPdbIdAndSelection("4cha", // and a collection of sequence positions to extract residues to use as motif List.of(new LabelSelection("B", "1", 42), // HIS new LabelSelection("B", "1", 87), // ASP new LabelSelection("C", "1", 47))) // SER .rmsdCutoff(1.0) .buildParameters() .buildContext() .run() .getHits() .stream() .map(hit -> hit.structureIdentifier() + "_" + hit.assemblyIdentifier() + " @ " + hit.labelSelections() + " - RMSD: " + hit.rmsd()) .forEach(System.out::println);This process can also be reversed to detect whether a structure of unknown function contains characteristic motifs.
// acquire a collection of motifs to screen for Set<EnrichedMotifDefinition> motifs = Strucmotif.getMotifDefinitionRegistry().getEnrichedMotifDefinitions(); Strucmotif.detectMotifs() .defineByPdbIdAndAssemblyId("2mnr", "1") .withMotifs(motifs) .rmsdCutoff(1.0) .buildParameters() .buildContext() .run() .getHits() .stream() .map(hit -> hit.motifIdentifier() + " @ " + hit.labelSelections() + " - RMSD: " + hit.rmsd()) .forEach(System.out::println);| Property | Action | Default Value/Behavior |
|---|---|---|
ccd-url | URL to the chemical component dictionary | wwPDB |
decimal-places-score | Number of decimal places reported for scores | 2 |
decimal-places-matrix | Number of decimal places reported in transformation matrices | 3 |
in-memory-strategy | Preload structure data for increased performance? | off |
loading-chunk-size | Batch size when holding structure data in memory | 200,000 |
max-results | Maximum number of results that will be returned | 50,000 |
max-motif-size | Maximum number of residues that may define a motif | 10 |
per-query-threads | Number of worker threads per query | available processors |
read-error-strategy | Behavior upon file bundle read error | exit |
query-timeout | Interrupt queries after n milliseconds | none |
root-path | Path where data files will be written | /opt/data/ |
Configure by placing your application.properties on the classpath. All properties specific to this project must be prefixed with strucmotif..
ccd-url provides a reference to the Chemical Component Dictionary. This file is used to handle non-standard amino acids appropriately if the modified-residue-strategy property is set to CCD_PARENT. This property controls how non-standard components are handled and the CCD provides a parent component for most modified residues, which can be used to map them to an amino acid that is known to the codebase (i.e., registered as ResidueType). Note that this property is both needed during the update and at runtime to resolve residue types.
You will need to process your corpus of structure data before using the service. This will create an optimized version of all structure files and add them to an inverted index that allows efficient searching.
Details can be found in: UPDATE.md
The update produces 5 files that are needed for the service to run:
| File | Format | Details |
|---|---|---|
| known.list | human-readable TSV | current holdings, incl. identifiers of all index entries and their revision history |
| renumbered.ffindex | human-readable TSV | summary of all optimized 3D structure data as .bcif.gz |
| renumbered.data | BinaryCIF files | all optimized 3D structure data, concatenated into one file, separated by \0 |
| index.ffindex | human-readable TSV | summary of all inverted index files (one per present residue-pair descriptor) |
| index.data | colfer files | all individual index files, concatenated into one file, separated by \0 |
Note that these files can't be mixed-and-matched. They contain cross-references and if you update or manipulate one, you'll need to edit all other files to ensure consistency. File bundles of .ffindex and .data can be read and manipulated using ffindex-java or similar implementations.
known.list contains tab-separated 4 values per line: entryId (original identifier), structureIndex (internal int identifier), majorRevision, and minorRevision (tracking the revision history and informing what needs updating).
.ffindex files contain 3 tab-separated values per line: filename, offset (long value that captures where this file starts), and length (filesize in bytes).
Entries might have prohibitively long String identifiers. The library uses an internally managed StructureIndex to address individual structures by a simple int value. This mapping is informed by the content of known.list and established by StructureIndexProvider.
Two address schemes exist. LabelSelection is a high-level, object-based way of referencing individual residues. It uses a combination of mmCIF properties, namely label_asym_id, struct_oper_id, and label_seq_id:
LabelSelection ref = new LabelSelection("A", "1", 123);Internally, access is facilitated using 32-bit unsigned primitive encoded integers (ResidueIndex). It doesn't follow any particular layout rather, all encountered residues are addressed by their index. Chain boundaries are ignored. Operations required for assemblies are honored as they occur in the source file and merely increment the counter. Additional work preserves information on chains and assemblies. Chain and operator names as well as boundaries are stored in memory and can be used to reconstruct LabelSelection instances if needed.
Residue pairs are identified by pairs of these int values. They can be stored as long value by chaining together 1st and 2nd value.
Residue pair descriptors capture the label_comp_id of both interacting residue, their backbone distance, their side-chain distance, and the angle defined between both.
They might look like AL-4-5-4, meaning that an alanine is in contact with a leucine, the distance of their CA atoms is in bin 4, i.e. [4.5, 5.5) A, the distance between their CB atoms is in bin 5, i.e. [5.5, 6.5) A, and the angle between both per-amino-acid vectors is in bin 4, i.e. [70, 90) deg.
These values are the Cartesian product of ResidueType (A, 36 states, 6 bits) x ResidueType (B, 36 states, 6 bits) x DistanceType (C, 32 states, 5 bits) x DistanceType (D, 32 states, 5 bits) x AngleType (E, 10 states, 4 bits) and are stored in an unsigned 32-bit integer. The 32-bit descriptors will use their 4th bit to store metadata (M) that tracks whether the identifier is flipped.
XXXMAAAA AABBBBBB XXCCCCCD DDDDEEEE A second flavor exists that only tracks DistanceType x DistanceType x AngleType and can be held in an unsigned 16-bit short.
XXCCCCCD DDDDEEEE Convenience functions to work with these descriptors are provided in the ResiduePairDescriptor class and provide ways to convert between the int representation and its human-readable form.
The inverted index is structured by residue pair descriptors. All residue pair descriptors with the same geometric properties (e.g., AL-4-5-4) are grouped together in one file.
Each file holds 3 pieces of data:
int[] structureIndices- references each entry that contains at least 1 occurrence of this residue pair (seeStructureIndexabove)int[] positionOffsets- defines boundaries between data on individual structuresint[] identifierData- pairs of residues within a specific structure of a certain residue pair descriptor (seeResidueIndexabove)
This data structure is encoded using colfer. This produces a lot of small-ish files. These files are bundled using ffindex-java.
- ciftools-java: mmCIF parsing and BinaryCIF implementation
- ffindex-java: bundle large amounts of small files together
- rcsb-molstar: define motifs in 3D and visualize results
Bittrich S, Burley SK, Rose AS (2020) Real-time structural motif searching in proteins using an inverted index strategy. PLoS Comput Biol 16(12): e1008502. https://doi.org/10.1371/journal.pcbi.1008502